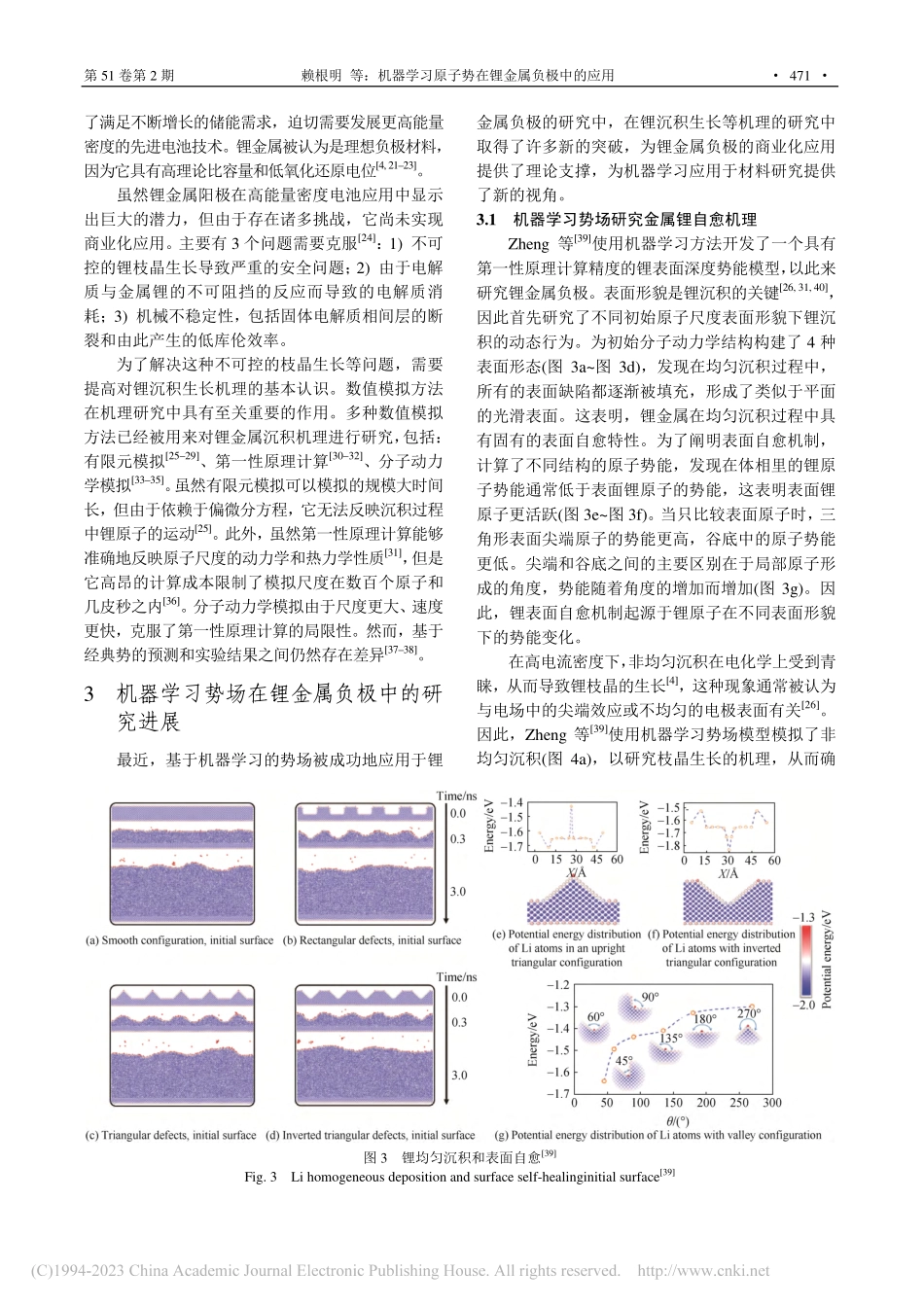
第51卷第2期2023年2月硅酸盐学报Vol.51,No.2February,2023JOURNALOFTHECHINESECERAMICSOCIETYhttp://www.gxyb.cbpt.cnki.netDOI:10.14062/j.issn.0454-5648.20220793机器学习原子势在锂金属负极中的应用赖根明1,焦君宇1,蒋耀2,郑家新1,2,欧阳楚英2(1.北京大学深圳研究生院新材料学院,广东深圳518055;2.宁德时代21C创新实验室,福建宁德352100)摘要:锂金属是下一代二次电池的理想负极材料。然而,锂枝晶生长存在安全隐患,并导致电池Coulombic效率低,这严重制约了锂二次电池的商业应用。目前,人们对锂的沉积生长机制在原子尺度上还了解甚少,同时对锂枝晶的成因也众说纷纭。近年来,机器学习在计算材料学中的应用使得许多以前无法实现的进步成为可能,本文综述了机器学习原子势在锂金属负极研究中的应用。关键词:锂金属负极;机器学习;分子动力学;计算模拟中图分类号:O646文献标志码:A文章编号:0454–5648(2023)02–0469–07网络出版时间:2022-12-28AnalysisofLiMetalAnodebyMachineLearningPotentialLAIGenming1,JIAOJunyu1,JIANGYao2,ZHENGJiaxin1,2,OUYANGChuying2(1.SchoolofAdvancedMaterials,PekingUniversity,ShenzhenGraduateSchool,Shenzhen518055,Guangdong,China;2.Fujianscience&technologyinnovationlaboratoryforenergydevicesofChina(21C-LAB),Ningde352100,Fujian,China)Abstract:Limetalisregardedasanidealanodeforthenext-generationsecondarybatteries.However,thegrowthofLi-dendriteresultsinalowcoulombefficiency,thusrestrictingthecommercialapplicationofLisecondarybatteries.ThemechanismofLidepositionandgrowthinanatomicscaleisstillunclear,andtherearedifferentopinionsabouttheoriginofLidendrite.Recentworkhasdealtwiththeapplicationofmachinelearningincomputationalmaterialsscience.ThisreviewrepresentedtheapplicationsofmachinelearningpotentialforthestudyofLimetalanode.Keywords:lithiummetalanode;machinelearning;moleculardynamics;simulation锂金属具有较低的电化学势(与标准氢气电极相比为–3.04V)和高达3860mA·h·g-1的比容量,使其成为下一代二次电池的理想负极材料[1–3]。然而,锂二次电池的商业化应用受到安全问题和低Coulombic效率的阻碍[4–5]:在循环过程中如果不受控制的锂枝晶生长穿透了隔膜,电池会短路[6],而形成的“死锂”会降低Coulombic效率[7]。多种表...