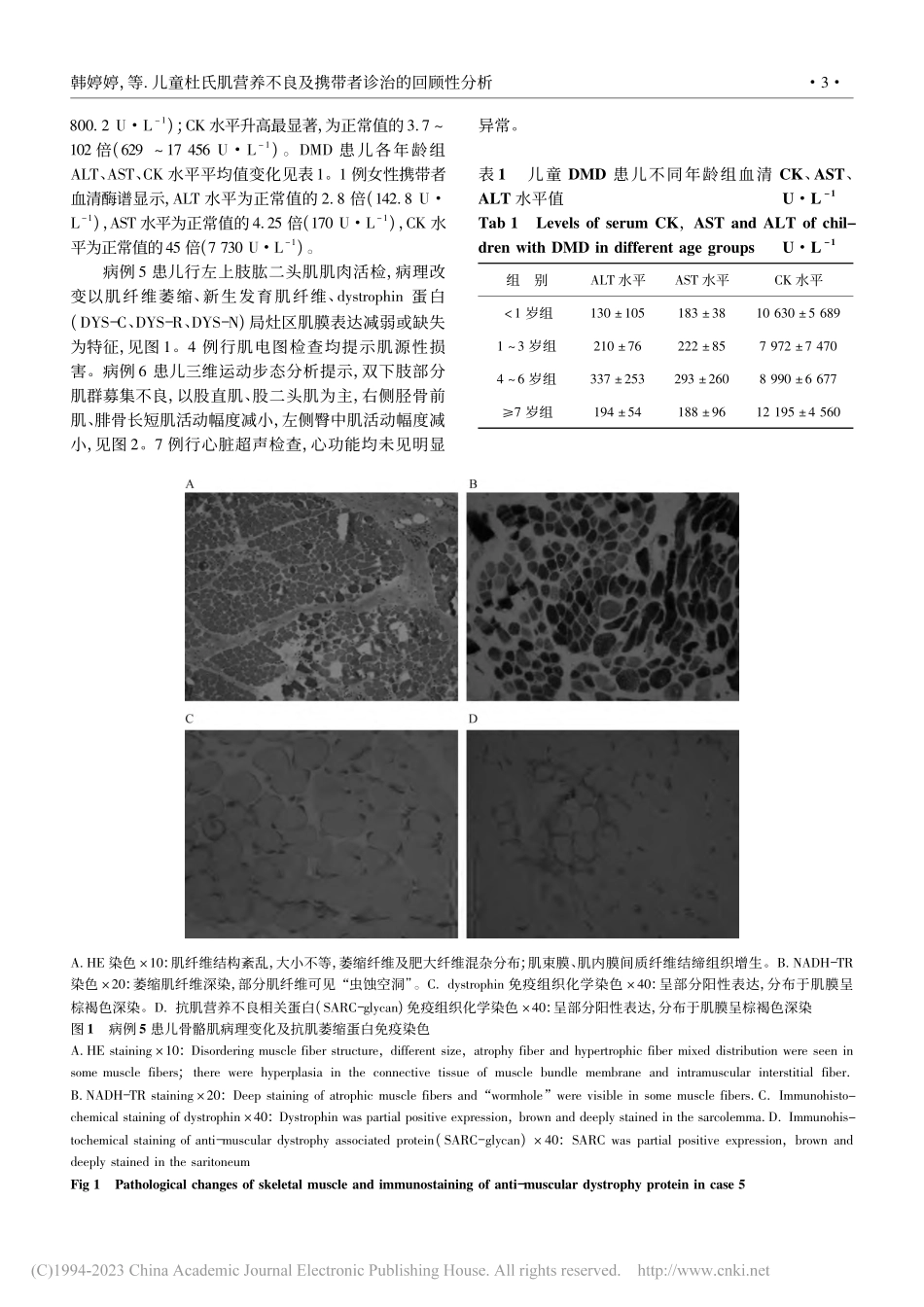
东南大学学报(医学版)JSoutheastUniv(MedSciEdi)2023,Feb;42(1):1-8[收稿日期]2022-10-14[修回日期]2022-11-09[基金项目]国家自然科学基金面上项目(81770162)[作者简介]韩婷婷(1987-),女,山西晋中人,主治医师,医学硕士。E-mail:hantingtingnetany@163.com[通信作者]李俊霞E-mail:lijunxia106@126.com[引文格式]韩婷婷,孙雪萍,陈辉,等.儿童杜氏肌营养不良及携带者诊治的回顾性分析[J].东南大学学报(医学版),2023,42(1):1-8.·论著·儿童杜氏肌营养不良及携带者诊治的回顾性分析韩婷婷1,孙雪萍2,陈辉1,陆超1,李俊霞1(南京医科大学第一附属医院1.儿科,2.生殖遗传检测中心,江苏南京210029)[摘要]目的:探讨杜氏肌营养不良(Duchennemusculardystrophy,DMD)患儿及携带者的临床特点、基因突变类型、治疗等,为早期诊断该病及进一步有效治疗进行总结。方法:回顾性分析2010年1月至2022年6月在本院经基因检测确诊的17例男性DMD患儿及1例女性携带者的临床资料。结果:17例DMD患儿平均诊断年龄5.9岁(1个月~12岁),1例女性携带者诊断年龄6岁。血清酶谱示谷丙转氨酶、谷草转氨酶、乳酸脱氢酶、α-羟丁酸脱氢酶、肌酸激酶同工酶、肌酸激酶(creatinekinase,CK)水平均升高,以CK水平升高最为显著,为正常值的3.7~102倍(629~17456U·L-1)。18例患儿中83.3%(15/18)存在缺失突变,其中45~55外显子缺失占41%;11.1%(2/18)为重复突变;5.5%(1/18)为内含子剪切突变。13例进行母亲基因检测验证,3例母亲未见DMD基因突变。1例患儿左上肢肱二头肌肌肉活检显示,病理改变为肌纤维萎缩、新生发育肌纤维、dystrophin蛋白局灶区肌膜表达减弱或缺失。4例肌电图检查均提示肌源性损害,DMD患儿三维运动步态分析提示双下肢部分肌群募集不良,以股直肌、股二头肌为主。1例DMD患儿行脐血间充质干细胞输注,5d后CK下降77.0%。结论:对血清肌酶异常、运动功能异常儿童应高度警惕DMD,尽早行基因检测确诊,家族女性携带者须进行遗传咨询和产前诊断。[关键词]杜氏肌营养不良;肌酸激酶;基因突变;间充质干细胞;儿童[中图分类号]R726.85[文献标志码]A[文章编号]1671-6264(2023)01-0001-08doi:10.3969/j.issn.1671-6264.2023.01.001AretrospectiveanalysisofdiagnosisandtreatmentofchildrenandcarrierwithDuchennemusculardystrophyHANTingting1,SUNXueping2,CHENHui1,LUChao1,LIJunxia1(1.DepartmentofPediatrics,2.D...