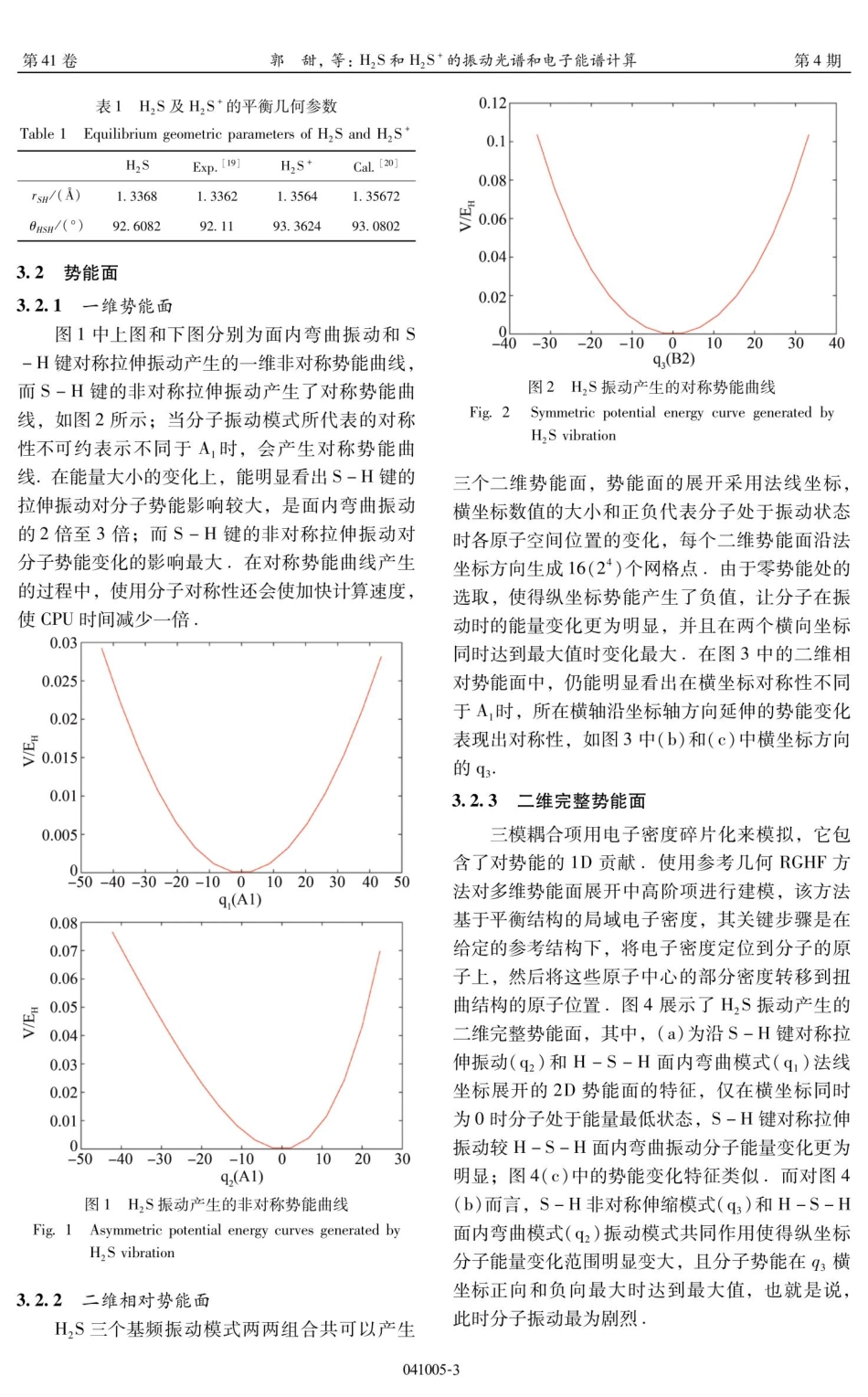
第41卷第4期2024年8月J.At.Mol.Phys.,2024,41:041005(8pp)原子与分子物理学报JOURNALOFATOMICANDMOLECULARPHYSICSH,S和H,S+的振动光谱和电子能谱计算Vol.41No.4Aug.2024郭甜,徐建刚,张佳美,张思雨,陈艳南,张云光(西安邮电大学理学院,西安710121)摘要:本文基于MOLPRO软件包使用从头算方法计算了星际分子H,S及其阳离子H,S+的势能面及光电子能谱:首先,在(U)CCSD/cc-pVQZ理论水平下获取了H,S沿法线坐标展开的势能面,势能面直观描述了不同振动模式耦合对分子能量变化的影响,S-H键的非对称伸缩振动和面内弯曲振动共同作用使得系统势能变化范围明显变大:振动多组态相互作用方法被用来计算非谐振动频率和振动光谱,计算结果显示,倍频和组合频之间出现了强烈的费米共振,使得相应波段处的红外强度显著增强最后,使用拉曼波函数和收缩不变Krylov子空间方法首次计算了H,SX'A一H,S+XB,的光电子能谱:此项研究有助于进一步理解星际分子的内部结构,并为实验研究及星际观测提供参考,关键词:势能面;VMRCI;振动光谱;光电子能谱中图分类号:0561VibrationalspectraandphotoelectronspectracalculationsofH,SandH,S文献标识码:AD0I:10.19855/j.1000-0364.2024.041005GUOTian,XUJian-Gang,ZHANGJia-Mei,ZHANGSi-Yu,CHENYan-Nan,ZHANGYun-Guang(SchoolofScience,Xi'anUniversityofPostsandTelecommunications,Xi'an710121,China)Abstract:ThepotentialenergysurfacesandphotoelectronspectraoftheinterstellarmoleculeH,SanditscationH,S+werecalculatedusingabinitiomethodsbasedontheMOLPROsoftwarepackage.First,thepotentialener-gysurfacesofH,Sexpandedalongthenormalcoordinateswereobtainedatthe(U)CCSD/cc-pVQZleveloftheory.Thepotentialenergysurfacesvisuallydescribetheeffectofdifferentvibrationalmodescouplingonthemolecularenergychange,andthecombinedeffectoftheasymmetricstretchingvibrationoftheS-Hbondandthein-planebendingvibrationmakesthepotentialenergychangerangeofthesystemsignificantlylarger.Thevibrationalmulti-referenceconfigurationinteractionmethodwasusedtocalculatetheanharmonicvibrationalfre-quenciesandvibrationalspectrum,andthecalculationsshowedthatstrongFermiresonancesappearbetweentheovertonesandcombinationbands,resultinginasignificantenhancementoftheinfraredintensityatthecorr...