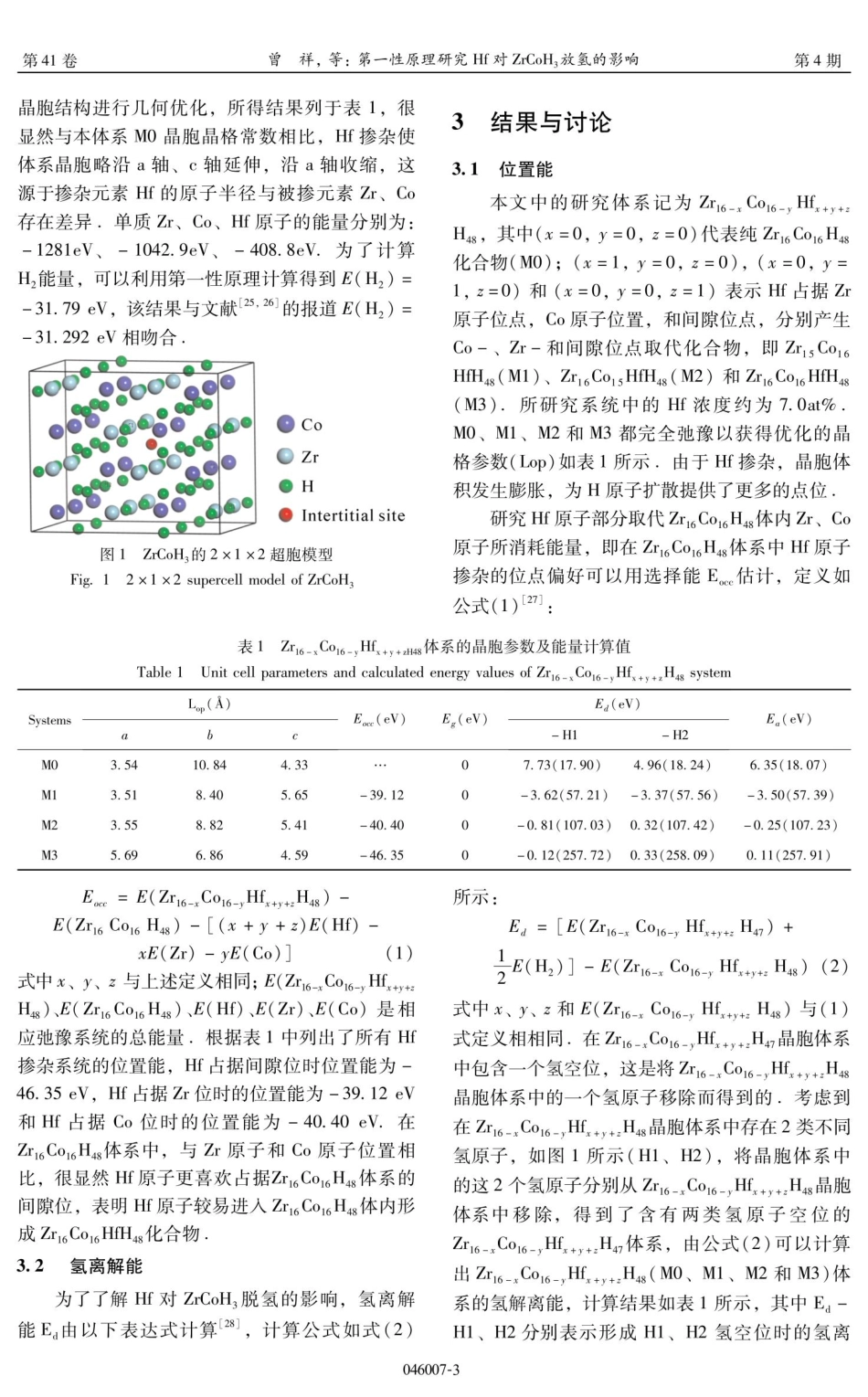
第41卷第4期2024年8月J.At.Mol.Phys.,2024,41:046007(9pp)原子与分子物理学报JOURNALOFATOMICANDMOLECULARPHYSICS第一性原理研究Hf对ZrCoH,放氢的影响Vol.41No.4Aug.2024曾祥,杨友山(铜仁学院农林工程与规划学院,铜仁554300)摘要:过渡金属元素Hf被设计成通过占据Co原子、Zr原子和间隙位点来添加到ZrCoH,中,通过第一性原理计算研究了Hf对ZrCoH,放氢的影响:发现用Hf掺杂会使ZrCoH,不稳定,导致氢离解能(Ea)、Co-H平均单位键长的键序(SBOco-H)降低,顺序为ZriCo6H4s>ZrieCo16HfH4s>ZricCoisHfH4s>ZrisCo16HfH4s·理论研究表明在ZrCoH,-Hf体系中,较弱的Co-H共价键相互作用、金属特性和Hf-H键的形成都有利于提高ZrCoH,的放氢能力.Hf原子优先占据间隙位,但这对氢离解能影响很小,氢离解能与位置能之间存在悖论关系,尽管ZrisCo16HfH4s化合物具有低的氢离解能,表现出良好的放氢性能,但在实际应用Hf占据Zr位时需要消耗过高的能量成本。关键词:储氢;ZrCoH;第一性原理计算;放氢性能;过渡金属元素掺杂中图分类号:K91Afirst-principlesstudyontheeffectofHfonthehydrogenevolutionofZrCoH,文献标识码:AD01I:10.19855/j.1000-0364.2024.046007ZENGXiang,YANGYou-Shan(SchoolofAgriculture,ForestryEngineeringandPlanning,TongrenUniversity,Tongren554300,China)Abstract:ThetransitionmetalelementHfisdesignedtobeaddedtoZrCoH,byoccupyingCoatoms,Zratomsandinterstitialsites.TheeffectofHfonthehydrogenevolutionofZrCoH,areinvestigatedbyfirst-principlescalculations.ItisfoundthatsubstitutionwithHfdestabilizesZrCoH3,leadingtoareductionsinthedehydrogen-ationenergy(Ed)andtheCo-HaveragebondlengthwiththeorderofZricCoicH4s>Zri6Co16HfH4s>ZriCo1sHfH4s>ZrisCoicHfH48.TheoreticalstudiesshowthatintheZrCoH,-Hfsystem,theweakerCo-Hcovalentbondinteraction,metallicpropertiesandtheformationofHf-HbondsareallbeneficialtoimprovethehydrogenreleasecapacityofZrCoH3.Hfatomspreferentiallyoccupyinterstitialsites,butthishaslittleeffectonthehydro-gendissociationenergy.AlthoughtheZrisCoicHfH4scompoundhaslowhydrogendissociationenergyandexhibitsgoodhydrogendesorptionperformance,itneedstoconsumetoohighenergycostwhenHfoccupiesZrsiteinpracticalapplication.Keywords:Hydrogenstorage;ZrCoH,;First-pri...