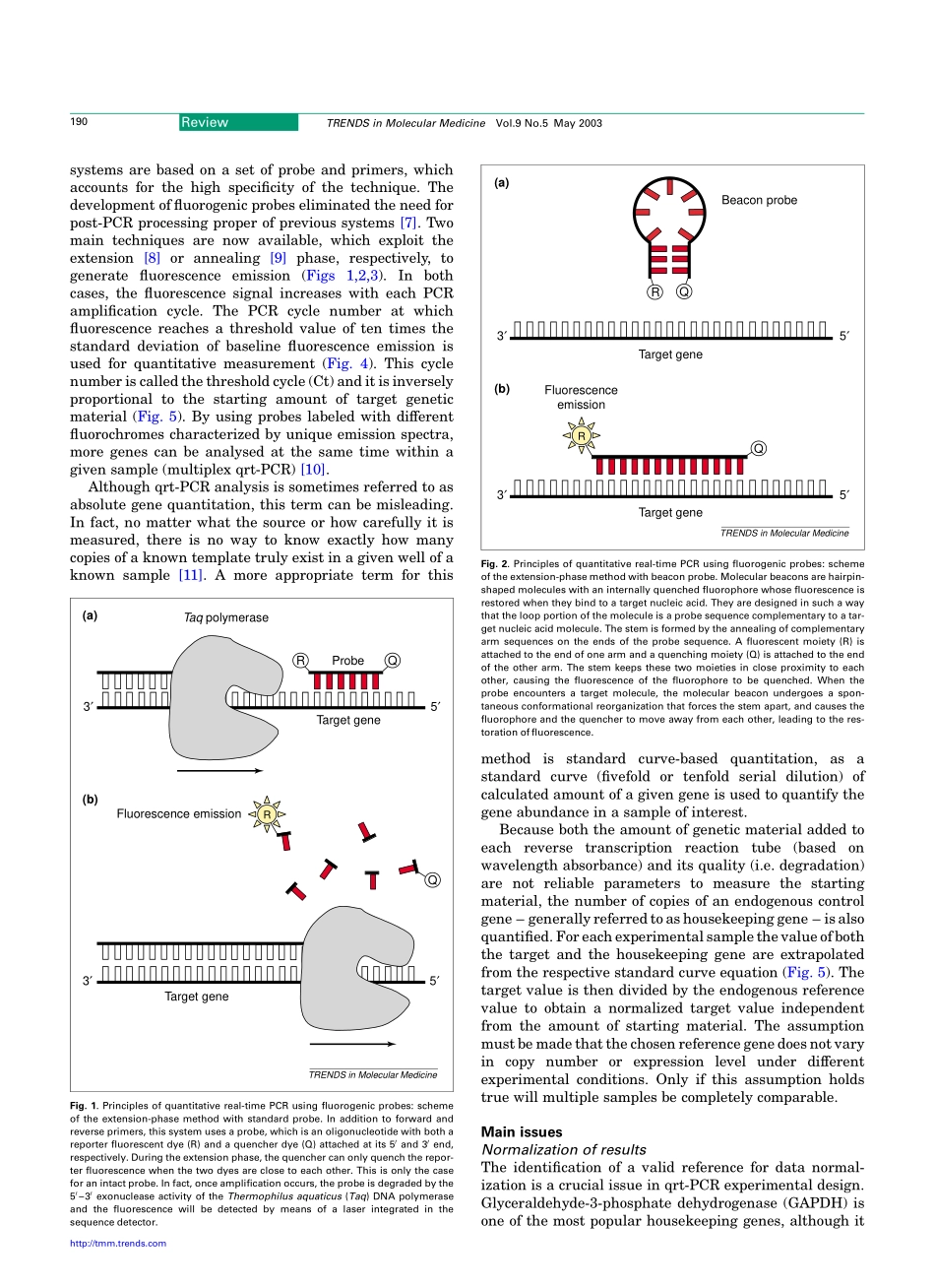
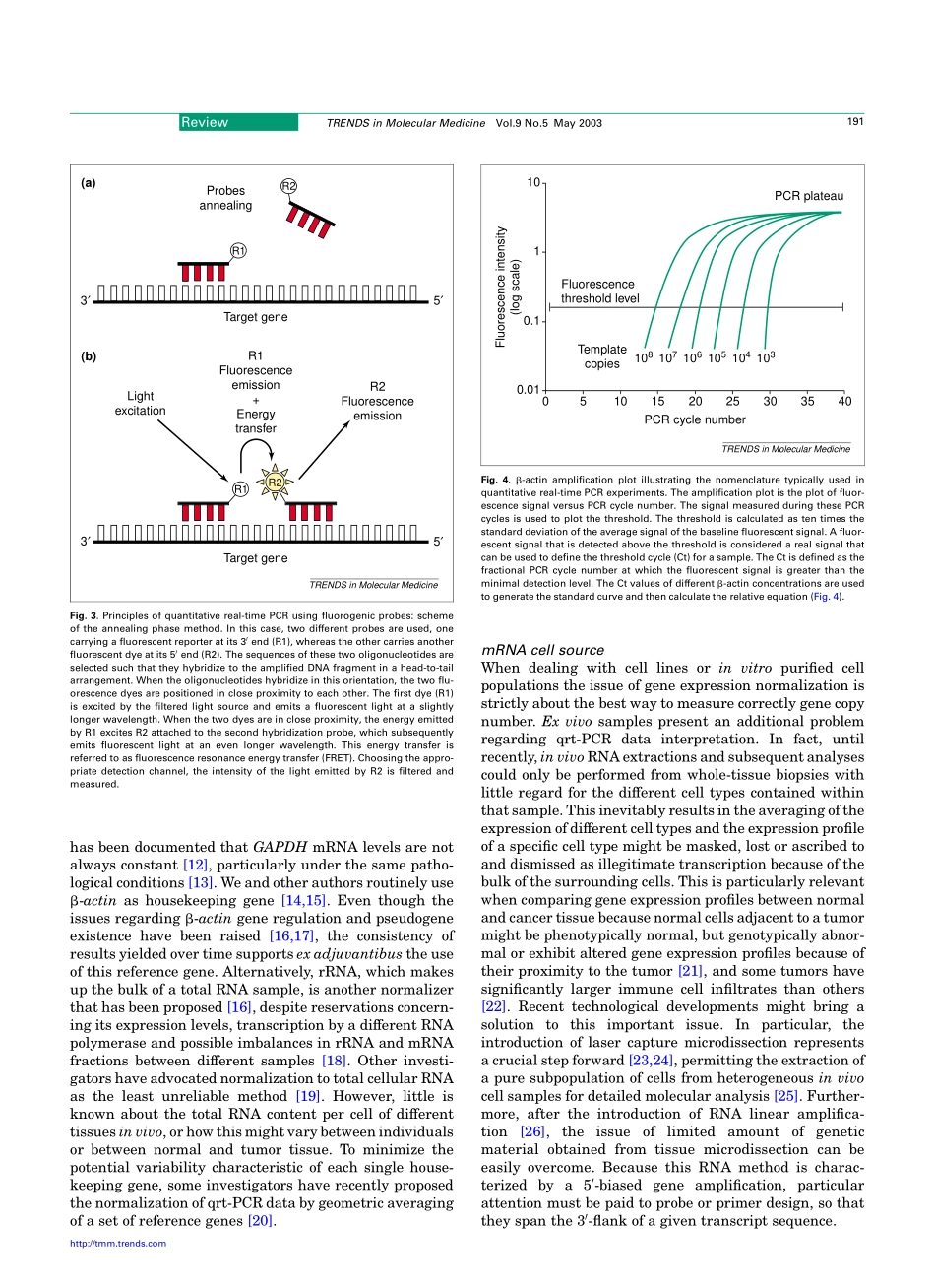
Quantitativereal-timePCR:apowerfulallyincancerresearchSimoneMocellin1,CarloR.Rossi1,PierluigiPilati1,DonatoNitti1andFrancescoM.Marincola21SurgeryBranch,DepartmentofOncologicalandSurgicalSciences,UniversityofPadova,viaGiustiniani2,35128Padova,Italy2ImmunogeneticsLaboratory,DepartmentofTransfusionMedicine,ClinicalCenter,NationalInstitutesofHealth,10CenterDrive,Bethesda,MD20814,USAInthiseraoftheHumanGenomeProject,quantitationofgeneexpressionintumororhostcellsisofpara-mountimportanceforinvestigatingthegenepatternsresponsibleforcancerdevelopment,progressionandresponseorresistancetotreatment.Quantitativereal-timePCR(qrt-PCR)technologyhasrecentlyreachedalevelofsensitivity,accuracyandpracticaleasethatsup-portsitsuseasaroutinebioinstrumentationforgenelevelmeasurement.Severalapplicationshavealreadybeenimplementedinthefieldofcancerresearch,andothersarebeingvalidated,showingthatthismolecularbiologytoolcanprovidebothresearchersandclinicianswithpreciousinformationconcerningthebehavioroftumors.Knowledgeofthebiochemicalprinciplesunderlyingthisbiotechnologycanbeofgreatvaluetointerpretcorrectlyqrt-PCRdata.PCR-basedtechniquesenableustoobtaingeneticinfor-mationthroughthespecificamplificationofnucleicacidsequences,startingwithaverylownumberoftargetcopies.Thesereactionsarecharacterizedbyalogarithmicamplificationofthetargetsequences;thatis,increaseofPCRcopiesfollowedbyaplateauphaseshowingarapiddecreasetozeroofcopynumberincrementpercycle.Accordingly,theamountofspecificDNAproductattheendofthePCRrunbearsnocorrelationwiththenumberoftargetcopiespresentintheoriginalspecimen.However,manyapplicationsinmedicineorresearchrequirequantificationofthenumberofspecifictargetsinthespecimenbothtostudythereactionofthecellorcellpopulationtoastimulusandtocomparethegeneprofileofdifferentsamples.AlthoughPCRanalysisgivesnoinformationonthebiologicallyactiveproductsofgenes(i.e.proteins),functionalgenomicsstudieshavedemon-stratedatightcorrelationbetweenthefunctionofaproteinandtheexpressionpatternsofitsgene[1].Th...