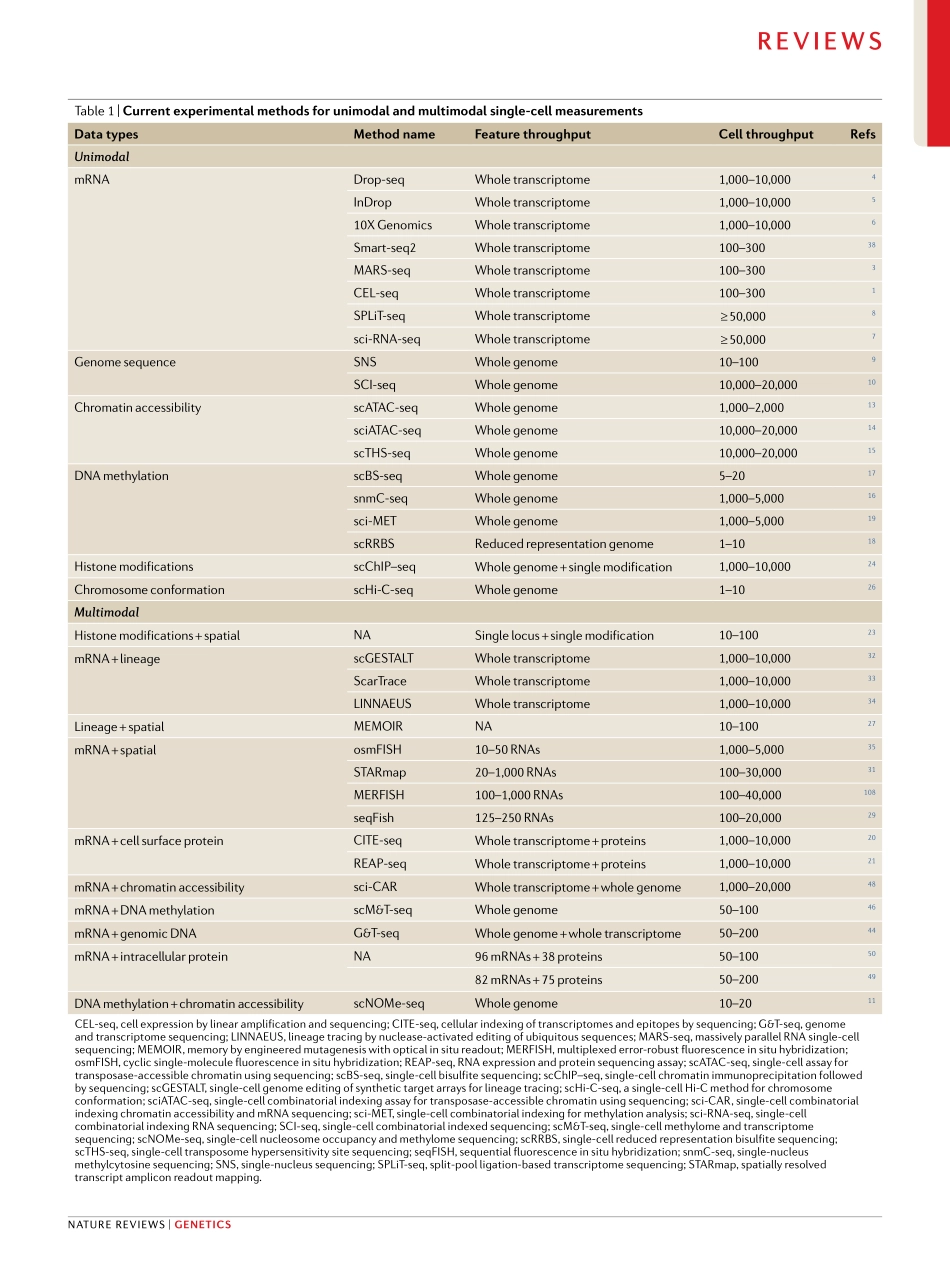
Recentadvancesinmolecularbiology,microfluidicsandnanotechnologyhavegivenrisetoamultitudeofsingle-cellsequencingtechnologies(Fig.1).Initialmeth-odshavefocusedonmeasurementsofasinglemodality(forexample,DNAsequence,RNAexpressionorchro-matinaccessibility).Althoughthesetechnologieshaveyieldedtransformativeinsightsintocellulardiversityanddevelopment,thissegregationisdrivenbymethodo-logicalconvenienceandlimitstheabilitytoderiveadeepunderstandingoftherelationshipsbetweenbiomole-culesinsinglecells.Understandingtheseinteractionsiskeytoderivingadeepunderstandingofthecellularstateandremainsachallengeforthefieldofsingle-cellanalysis.Moreover,asthescaleandavailabilityofdatasetsrapidlygrow,newcomputationalmethodsareneededfornormalizationandjointanalysisacrosssam-ples,eveninthepresenceofsignificantbatcheffectsorinterindividualvariation.Single-cellRNAsequencing(scRNA-seq)isoneofthemostwidelyusedsingle-cellsequencingapproaches,witharangeoftechnologiesforsensitive,highlymulti-plexedorcombinatoriallybarcodedprofiling1–8.Theseadvanceshaveaccompaniedavarietyofcomplementarysingle-cellgenomic,epigenomicandproteomicprofilingtechnologies,includingmethodsforsingle-cellmeasure-mentsofgenomesequence9,10,chromatinaccessibil-ity11–15,DNAmethylation11,16–19,cellsurfaceproteins20,21,smallRNAs22,histonemodifications23,24andchromo-somalconformation25,26.Furthermore,recenteffortshavepioneeredmethodstoaccuratelyrecordspatialorlineageinformationinsingle-cellstudies27–35(Fig.1;TAble1).Anidealizedexperimentalworkflowwouldobserveallaspectsofthecell,includingafullhistoryofitsmolecularstates,spatialpositionsandenvironmentalinteractions.Althoughoutsidetheboundsofcurrenttechnology,multimodaltechnologiesandintegrativecomputationalmethodsenableustomoveclosertothisaspirationalandexcitinggoal.InthisReview,wedescribethecurrentlyavailablemethodsforsingle-celltranscriptomics,genomics,epigenomicsandproteom-icswithanemphasisonthosemethodsthatprovidemultimodaldataordatathatcanbeintegratedintoamultimodalanalysis.W...