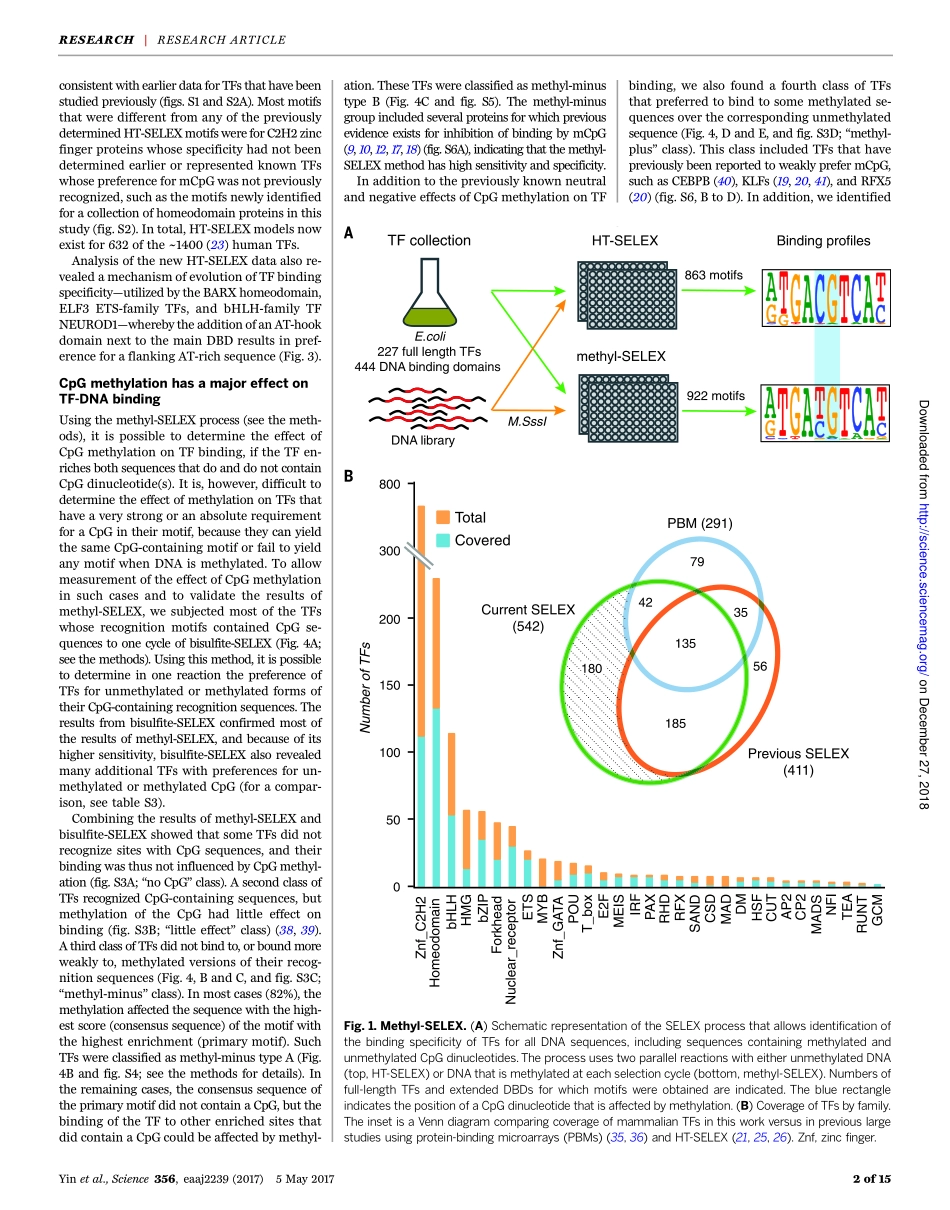
RESEARCHARTICLESUMMARY◥DNAMETHYLATIONImpactofcytosinemethylationonDNAbindingspecificitiesofhumantranscriptionfactorsYimengYin,EkaterinaMorgunova,ArttuJolma,EeviKaasinen,BiswajyotiSahu,SyedKhund-Sayeed,PratyushK.Das,TeemuKivioja,KashyapDave,FanZhong,KazuhiroR.Nitta,MinnaTaipale,AlexanderPopov,PaulA.Ginno,SilviaDomcke,JianYan,DirkSchübeler,CharlesVinson,JussiTaipale*INTRODUCTION:Nearlyallcellsinthehumanbodysharethesameprimarygenomesequenceconsistingoffournucleotidebases.Oneofthebases,cytosine,iscommonlymodifiedbymeth-ylationofits5positioninCpGdinucleotides(mCpG).MostCpGdinucleotidesinthehumangenomearemethylated,butthelevelofCpGmethylationvarieswithgeneticlocation(pro-moterversusgenebody),whethergenesareactiveversussilenced,andcelltype.ResearchhasshownthatthemaintenanceofaparticularcellularstateaftercelldivisionisdependentonfaithfultransmissionofmethylatedCpGs,aswellasinheritanceofthemothercells’rep-ertoireoftranscriptionfactorsbythedaughtercells.Thesetwomechanismsofepigeneticinher-itancearelinkedtoeachother;thebindingoftranscriptionfactorscanbeaffectedbycy-tosinemethylation,andcytosinemethylationcan,inturn,beaddedorremovedbyproteinsthatassociatewithtranscriptionfactors.RATIONALE:Thegeneticandepigeneticlan-guage,whichimpartswhenandwheregenesareexpressed,isunderstoodataconceptuallevel.However,amoredetailedunderstandingisneededofthegenomicregulatorymechanismbywhichmethylatedcytosinesaffecttranscrip-tionfactorbinding.BecausecytosinemethylationchangesDNAstructure,ithasthepotentialtoaffectbindingofalltranscriptionfactors.However,asystematicanalysisofbindingofalargecollectionoftranscriptionfactorstoallpossibleDNAsequenceshasnotpreviouslybeenconducted.RESULTS:Togloballycharacterizetheeffectofcytosinemethylationontranscriptionfactorbinding,wesystematicallyanalyzedbindingspecificitiesoffull-lengthtranscriptionfactorsandextendedDNAbindingdomainstoun-methylatedandCpG-methylatedDNAbyusingmethylation-sensitiveSELEX(systematicevo-lutionofligandsbyexponentialenrichment).Weevalu...