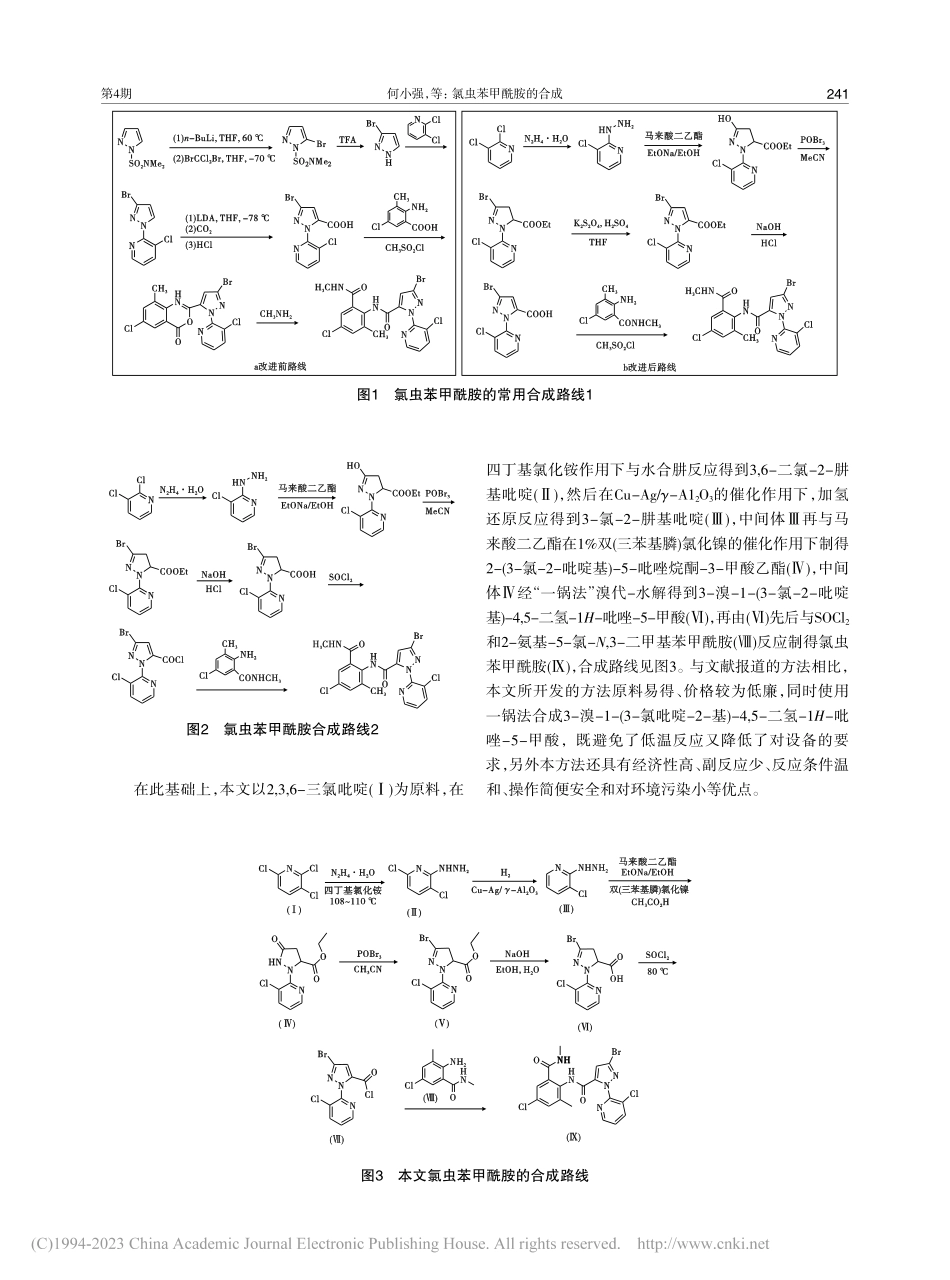
农药AGROCHEMICALS何小强,陈志忠,张黎,等.氯虫苯甲酰胺的合成[J].农药,2023,62(4):240-243.doi:10.16820/j.nyzz.2023.4001收稿日期:2022-10-16,修返日期:2022-11-18作者简介:何小强(1983—),男,四川南充人,高级工程师,主要从事吡啶及其衍生物类化工中间体的相关研究。E-mail:642040863@qq.com。通讯作者:李倩文(1994—),女,新疆伊犁州人,工程师,主要从事精细化工相关的研究。Tel:023-61000631,E-mail:1575671316@qq.com。氯虫苯甲酰胺的合成何小强,陈志忠,张黎,芮正军,胡娜,李倩文(重庆华歌生物化学有限公司,重庆404000)摘要:[目的]开发出一种反应条件温和、原料成本低、适合工业化生产的氯虫苯甲酰胺的合成方法。[方法]以廉价的2,3,6-三氯吡啶为原料,经肼解、加氢还原、环合、“一锅法”溴代-水解得到3-溴-1-(3-氯-2-吡啶基)-4,5-二氢-1H-吡唑-5-甲酸,然后分别与SOCl2和2-氨基-5-氯-N,3-二甲基苯甲酰胺反应得到氯虫苯甲酰胺。[结果]该方法合成的产物含量为95.5%,收率为51.09%(以原料2,3,6-三氯吡啶计)。[结论]所开发的方法既避免了反应条件苛刻和环境不友好等缺点,又降低了合成成本。关键词:氯虫苯甲酰胺;2,3,6-三氯吡啶;催化剂;合成中图分类号:TQ460.3文献标志码:A文章编号:1006-0413(2023)04-0240-04ThesynthesisofchlorantraniliproleHEXiao-qiang,CHENZhi-zhong,ZHANGLi,RUIZheng-jun,HUNa,LIQian-wen(ChongqingHuageBiochemistryLimitedCompany,Chongqing404000,China)Abstract:[Aims]Thisstudyaimstodevelopasyntheticmethodofchlorantraniliprolewithmildreactionconditions,lowrawmaterialcostandsuitableforindustrialproduction.[Methods]Usinglow-cost2,3,6-trichloropyridineastherawmaterial,3-bromo-1-(3-chloro-2-pyridyl)-4,5-dihydro-1H-pyrazole-5-carboxylicacidwasobtainedbyreactionsofhydrazinolysis,hydrogenationreduction,cyclizationand"onepot"bromination-hydrolysis,whichreactedwithSOCl2and2-amino-5-chloro-N,3-dimethylbenzamiderespectivelytodeliverchlorantraniliproel.[Results]Thecontentoftheproductsynthesizedbythismethodwas95.5%,andtheyieldwas51.09%(basedontherawmaterial2,3,6-trichloropyridine).[Conclusions]Themethodavoidsthedisadvantagesofharshreactionconditionsandenvironmentunfriendliness,andreducesthesynthesiscost.Keywords:chlorantranilipr...