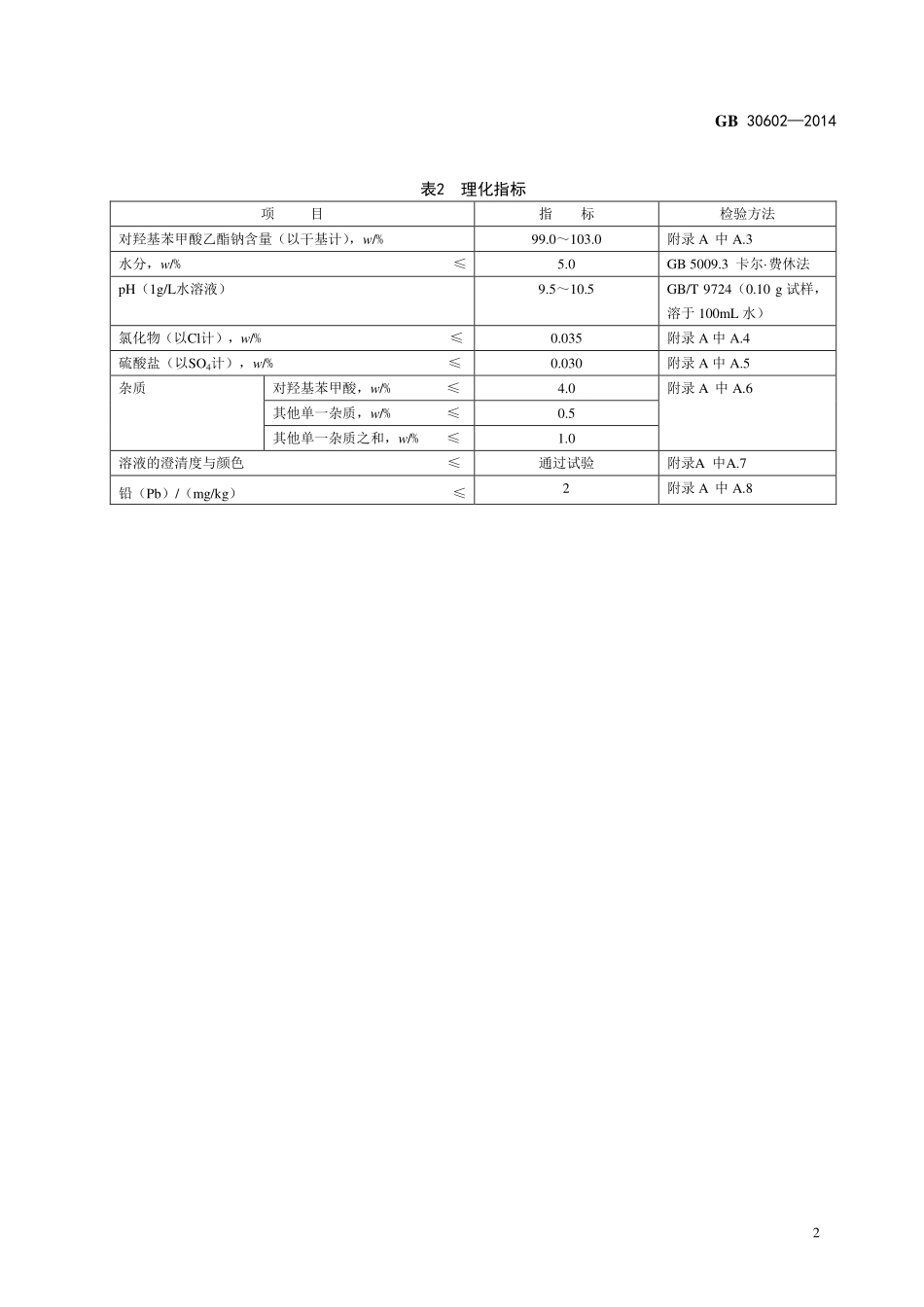
中华人民共和国国家标准GB30602—2014食品安全国家标准食品添加剂对羟基苯甲酸乙酯钠2014-04-29发布2014-11-01实施GB30602—20141食品安全国家标准食品添加剂对羟基苯甲酸乙酯钠1范围本标准适用于在氢氧化钠水溶液中加入对羟基苯甲酸乙酯反应后精制而成的食品添加剂对羟基苯甲酸乙酯钠。2化学名称、分子式、结构式和相对分子质量2.1化学名称对羟基苯甲酸乙酯钠2.2分子式C9H9NaO32.3结构式2.4相对分子质量188.2(按2007年国际相对原子质量)3技术要求3.1感官要求:应符合表1的规定。表1感官要求项目要求检验方法色泽白色或近白色取适量试样,置于清洁、干燥的白瓷盘中,在自然光线下,观察其色泽和状态状态粉末3.2理化指标:应符合表2的规定。GB30602—20142表2理化指标项目指标检验方法对羟基苯甲酸乙酯钠含量(以干基计),w/%99.0~103.0附录A中A.3水分,w/%≤5.0GB5009.3卡尔·费休法pH(1g/L水溶液)9.5~10.5GB/T9724(0.10g试样,溶于100mL水)氯化物(以Cl计),w/%≤0.035附录A中A.4硫酸盐(以SO4计),w/%≤0.030附录A中A.5杂质对羟基苯甲酸,w/%≤4.0附录A中A.6其他单一杂质,w/%≤0.5其他单一杂质之和,w/%≤1.0溶液的澄清度与颜色≤通过试验附录A中A.7铅(Pb)/(mg/kg)≤2附录A中A.8GB30602—20143附录A检验方法A.1一般规定除非另有说明,在分析中仅使用确认为分析纯的试剂和GB/T6682中规定的三级水。分析中所用标准滴定溶液、杂质标准溶液、制剂及制品,在没有注明其他要求时,均按GB/T601、GB/T602和GB/T603之规定制备。试验中所用溶液在未注明用何种溶剂配制时,均指水溶液。A.2鉴别试验A.2.1试剂和材料A.2.1.1盐酸。A.2.1.2盐酸溶液:1+1。A.2.1.3碳酸钠溶液:106g/L。A.2.1.4铁氰化钾溶液:53g/L。A.2.1.54-氨基安替比林-硼酸盐缓冲溶液:0.1%。量取含0.618%硼酸的0.1mol/L氯化钾溶液1000mL与0.1mol/L氢氧化钠溶液420mL混合,得到pH9.0的硼酸盐缓冲溶液。取1g4-氨基安替比林溶于1000mL硼酸盐缓冲溶液中。A.2.1.6乙酸氧铀锌溶液。称取10g乙酸双氧铀,加5mL冰乙酸和50mL水,微热使溶解;称取30g乙酸锌,加3mL冰乙酸和30mL水,微热使溶解;将两溶液混合,放冷,过滤,即得。A.2.2鉴别步骤A.2.2.1对羟基苯甲酸乙酯的熔点称取0.5g试样,精确至0.01g,加入50mL水使溶解,立即加5mL盐酸,振摇,过滤,用水充分洗涤沉淀,在80℃下减压干燥2h。按GB/T617测定试样熔点,熔点为115℃~118℃。A.2.2.2显色反应取0.01g试样,置于试管中,加1m...