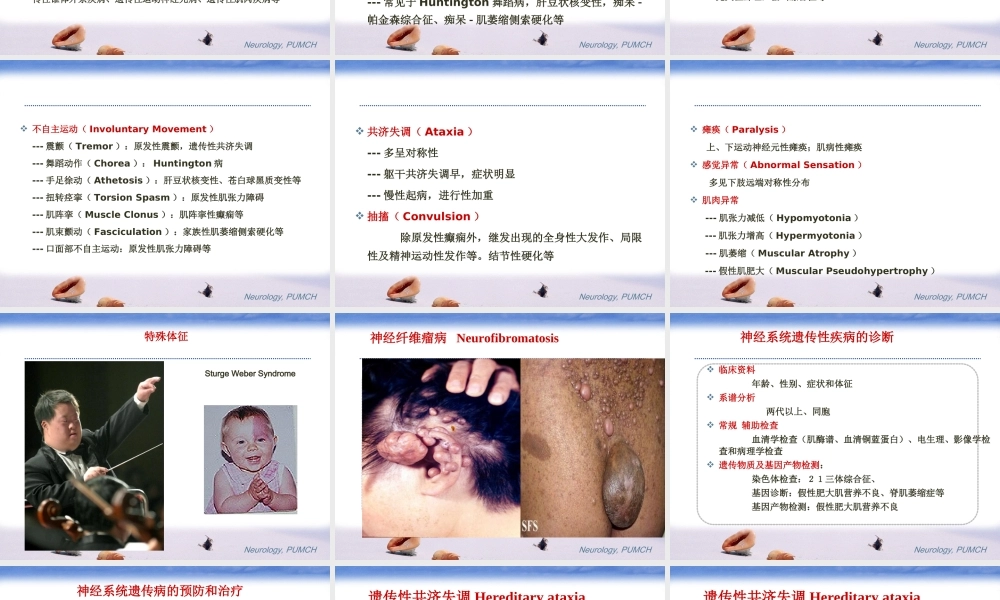
神经系统遗传性疾病北京协和医院神经科彭斌Neurology,PUMCH简介先天性疾病、家族性疾病(单个家族?多个家族?环境因素)神经系统遗传病:居各系统遗传病之首,60%以上,国内神经系统单基因遗传病患病率109.3/10万,以遗传性共济失调和进行性肌营养不良症最常见发病年龄:任何年龄均可发病,与疾病类型有关Neurology,PUMCH神经系统遗传疾病的分类根据遗传物质改变分类:一、单基因遗传病:---常染色体显性遗传:神经纤维瘤病、结节性硬化、腓骨肌萎缩等。---常染色体隐性遗传:肝豆状核变性、苯丙酮尿症---X连锁隐性遗传:男性发病,假肥大型肌营养不良---X连锁显性遗传:某些腓骨肌萎缩症二、多基因遗传病:癫痫、帕金森病、老年性痴呆Neurology,PUMCHNeurology,PUMCH三、线粒体遗传病:线粒体肌病、线粒体脑肌病等四、染色体病:21三体综合征(先天愚型)根据病变的部位分类:遗传性周围神经病、遗传性脊髓-脑干-小脑系统疾病、遗传性锥体外系疾病、遗传性运动神经元病、遗传性肌肉疾病等Neurology,PUMCH神经系统遗传病的症状学智能发育不全(MentalRetardation)---精神发育迟滞:重度(白痴,智商0~30)中度(痴愚,智商30~50)轻度(鲁钝,智商50~70)见于大多数常染色体病,遗传代谢病痴呆(Dementia)---常见于Huntington舞蹈病,肝豆状核变性,痴呆-帕金森综合征、痴呆-肌萎缩侧索硬化等Neurology,PUMCH行为异常(AbnormalBehavior)---发作性兴奋,冲动,易激惹,烦躁不安,攻击行为,人格行为异常,如自毁容貌等。语言障碍(SpeechDisorder)---智能障碍多伴有语言障碍---发音障碍:痉挛性发音障碍、弛缓性发音障碍、运动失调性发音困难、运动障碍性发音困难---先天性聋哑,猫叫综合征等Neurology,PUMCH不自主运动(InvoluntaryMovement)---震颤(Tremor):原发性震颤,遗传性共济失调---舞蹈动作(Chorea):Huntington病---手足徐动(Athetosis):肝豆状核变性、苍白球黑质变性等---扭转痉挛(TorsionSpasm):原发性肌张力障碍---肌阵挛(MuscleClonus):肌阵挛性癫痫等---肌束颤动(Fasciculation):家族性肌萎缩侧索硬化等---口面部不自主运动:原发性肌张力障碍等Neurology,PUMCH共济失调(Ataxia)---多呈对称性---躯干共济失调早,症状明显---慢性起病,进行性加重抽搐(Convulsion)除原发性癫痫外,继发出现的全身性大发作、局限性及精神运动性发作等。结节性硬化等Neurology,PUMCH瘫痪(Paral...